糖原贮积病
别名:糖原病概述
糖原贮积症(glycogen storage disease,GSD)是一组由糖原分解过程中酶缺乏引起的疾病。糖原是由许多葡萄糖组成的带分支的大分子多糖,主要存在于肝脏和肌肉中。糖原的分解过程是涉及多种酶的复杂酶促反应,其中任何一种酶的缺乏均可致病。目前糖原贮积症可分为13型,此类疾病发病机制可以1型(von Gierke病)为例说明。本病是由于肝内葡萄糖-6-磷酸酶缺乏引起,在新生儿和婴儿早期,有易激怒、苍白、发绀、喂养困难及低血糖抽搐、肝大等。患儿5~6岁后以出血、感染为主要症状。本病为常染色体隐性遗传,葡萄糖-6-磷酸酶基因定位于17号染色体。
病因

病因现已阐明,本病是由于糖原代谢过程中底物的累积所致。一般情况下,在酶促反应中,底物A经过酶x的催化作用生成产物B,产物B可能是另一个酶Y的底物。由于Y酶的作用,B转化为产物C,这种反应可以延续直到终末产物不能继续分解为止。
如果编码酶x的基因发生突变,酶x会丧失活性,这样底物A会在人体内累积。以底物的化学特性及累积的器官而异,患者可以产生不同的病症,这种遗传代谢疾病大多数是属于常染色体隐性遗传,糖原贮积症就是一个典型的范例。
症状
本型患者发病年龄和临床表现变异性较大。重症患儿在新生儿期即可出现严重低血糖症、高脂血症、酸中毒、呼吸困难、肝脏肿大和肾肿大等表现;轻症病例则常在幼儿期因生长迟缓、腹部膨胀等就诊。由于慢性乳酸酸中毒和长期胰岛素/胰高糖素比例失常,患儿身材明显矮小,骨龄落后,骨质疏松。肝脏持续增大可导致腹部显著隆起。肌肉松弛,四肢伸侧皮下常有黄色脂肪瘤。但身体各部分比例和智能发育可正常。患儿时有低血糖发作和腹泻发生。少数幼婴在重症低糖时可伴发惊厥或癫痫发作,但也有血糖降至0.56mmol/L以下而无明显症状者,随着年龄的增长,低血糖发作次数可以减少。由于血小板功能不良,患儿有鼻出血等出血倾向。
并发症
患者成年后累患心血管疾病、胰腺炎和肝腺瘤(或腺瘤)等机会增加。
诊断
1.肝型糖原贮积症的诊断:诊断线索为婴儿和儿童肝大,生长迟缓及频发低血糖。肝脏活组织检查示糖原累积,肝细胞内沉积的糖原用过碘酸-Schiff试剂染色呈强阳性,且病程早期即用淀粉酶治疗后这种反应会消失。在许多病例,胰高血糖素刺激试验后血糖升高低于正常(>2mmol/L);然而活检明确诊断对估测预后和指导治疗意义重大。肝组织的直接糖原和脂肪含量分析以及酶学检测是必要的。糖原的组织化学和电子显微镜研究可为诊断提供有用的信息。如果可行,应该开展肝脏的开放式楔形活检以取得足够的组织确定诊断。活检前,可以输注适量的血小板和凝血因子来纠正出血体质,并在直视下确保止血。然而,这种手术对酸中毒或出血倾向的幼儿是有危险的,应该密切观察防止低血糖的发生。
对于工型糖原贮积症的分型诊断尤其困难。葡萄糖-6-磷酸酶系统仅存在于内质网,有些成分结合在内质网膜上,因此,当将冷冻组织融化进行分析时,不可能诊断这些影响底物或产物转运的特定的损伤。诊断IB和IC型糖原贮积症(葡萄糖-6-磷酸移位酶缺陷),只能用新鲜组织分析。原因是冷冻-融化的组织微粒体酶系统的完整性被破坏,使其对磷酸盐、焦磷酸盐和葡萄糖-6-磷酸通透,从而掩盖了转运缺陷。因此,当怀疑有糖原贮积症时,应该选择有条件的实验室,可以用新鲜的和深度冷冻的活检组织进行检测。
2.肌型糖原贮积症的诊断:骨骼肌的化学能量供应通路为分解葡萄糖和糖原生成乳酸产生ATP,前臂运动试验对诊断这一通路缺陷有意义。无氧的条件下,糖酵解是产生ATP的唯一途径,糖酵解优先分解从糖原分解的葡萄糖,而不是从血浆摄取的葡萄糖。糖酵解的缺陷(Ⅶ型糖原贮积症和其他酶缺陷)会产生相似的症状。嘌呤通路障碍、卟胆色素原脱氨酶缺乏症的病人可能发生运动诱发的痉挛。可以通过运动试验安全地诊断这些疾病。与McArdle(1951)设计的早期试验不同,这些种激发试验不会导致骨骼肌溶解,血清肌酸激酶升高,最终导致肌红蛋白尿性肾衰竭。病史中的这些特征可能提示肌型糖原贮积症。
肌型糖原贮积症的确诊需要活检以进行组织化学、超微结构和生物化学分析。为与其他肌病鉴别,如肢带型肌营养不良、Duchenne肌营养不良、Kugelberg-Welander病(少年型家族性进行性脊肌萎缩症)、肌营养不良性肌强直症和线粒体肌病及继发性肌病,如多肌炎,需要做活检与肌电图。
前臂运动试验具体试验方法为:病人休息30min,从非运动的一侧手臂抽取肘静脉血。在另一侧手腕缠好一个血压计袖带。在这侧胳膊上端用另一个标准袖带充气到平均动脉压水平,让患者在2min内用最大力量握拳120次。然后,将手腕袖带马上充气到200mmHg。运动后2min,从运动侧手臂抽取肘静脉血,松开上面的袖带。每隔1min再抽取一次血,共取5个样本。样本应立刻送去实验室分析乳酸和氨含量。乳酸生成量下降或无乳酸生成是肌肉糖原和糖分解缺陷的特征。在Ⅲ、V和Ⅷ型糖原贮积症患者,血浆中氨(包括肌苷和次黄嘌呤)的水平明显升高。这些异常反映了ATP生成障碍的患者的运动肌肉中,嘌呤被过度降解。测定氨的水平也对运动试验的乳酸测定提供了鉴别价值,可以排除由于运动强度不足造成的乳酸水平低。此试验也可以鉴别卟胆色素原脱氨酶缺乏症,此病患者乳酸生成是正常的,但由于嘌呤循环的利用障碍和缺乏不能利用其作为替代底物生成能量,导致静脉血氨水平不升高。
3.分型诊断:Dunger等提出的对糖原贮积症分型的筛选的方案是有用的(图1)。
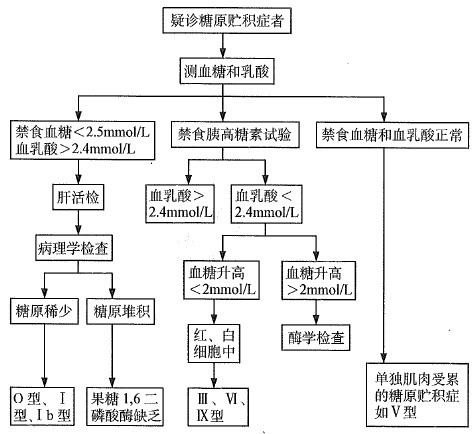
图1:糖原贮积病分型筛选
根据上述筛选方案,然后再做酶活性测定,这样可缩小酶活性测定的范围。
Ding等用免疫印染分析法测定了Ⅲ型糖原贮积症的酶活性(包括转移酶和α-糖苷酶)。此方法的原理是从病人活检所得组织制成匀浆作为抗原,与从猪肌肉中纯化的脱枝酶所制备的多克隆抗体作Western印染分析,可以将ⅢA、ⅢB和ⅢD型病人鉴定出来;用培养的羊水细胞可对胎儿可作出生前诊断。
Ⅰ型糖原贮积症的G6P活性的测定的原理是测定每克蛋白,每分钟释放出来的磷离子,1U的G6P活性是每分钟释放1μmol的磷离子。因为G6P在微粒体内,微粒体膜是限制性膜,具有选择性通透性。微粒体膜破裂后才有G6P活性表达。故测定G6P活性需测完整的和膜破裂后微粒体每分钟释放出来的磷离子的量,分别以IM(intact microsome)和DM(disruptive microsome)表示,同时要测定像G6P一样只在微粒体破裂后才有活性表达的甘露糖-6-磷酸和焦磷酸酶和蛋白质浓度。测定IM和DM状态下G6P的比例,然后再通过公式计算出完整微粒体中G6P的活性。测定前要分离出活检组织匀浆中的微粒体。Stamm等测定1例成年部分性G6P缺乏的病人结果为每克蛋白含2.4和1.98μmolpi/min,磷酸酶31.2μmolpi/min(正常对照者分别4.7±1.9和(25.1±6.5)μmolpi/min)。
各种类型的糖原贮积症中的酶活性均可测定,其方法各异,这里不作逐个介绍。总之,酶活性测定可确定糖原贮积症的类型。正是由于酶活性测定进行分型步骤繁杂、费力、费时,因此,有些学者提出可以DNA为基础的方法对怀疑患有糖原贮积病ⅠA型来进行初步筛查,可免除做肝活检和酶学诊断。
具有周围中性和单核白细胞减少或功能异常的Ⅰ型糖原贮积症中的ⅠB型和Ⅰc型可用周围中性白细胞做试验以检出ⅠB型病人酶的功能,并可与ⅠA型鉴别。ⅠB型病人的中性白细胞在加入葡萄糖时,NADPH氧化酶几乎无增加,而ⅠA型病人则明显增加。这是因为ⅠB型病人的中性白细胞缺乏对细胞外葡萄糖反应与加入葡萄糖不能提高细胞内G6P水平有关,由此限制了在己糖-1-磷酸旁路中NADPH的产生。此试验对确定ⅠB型病人的中性白细胞是否有功能异常是有力证据。
4.病因诊断(基因诊断):各种类型的糖原贮积症都是由糖原合成或糖原分解过程中某种酶缺如或活性减低。这些酶缺陷与酶的相关基因发生突变有关,只有极少数某种类型的糖原贮积症病人中未检出有相关基因突变。
检查基因突变都是采用分子生物学方法。常用方法为以多聚酶链式反应(PCR)为基础,即在提取酶基因DNA,对酶的所有外显子或个别外显子或所有外显子和内含子进行PCR扩增,然后对扩增后的产物进行DNA测序,或做单链构象多态性(SSCP),或做限切酶长度多态性(RELP)分析以检出突变。最近报道用TaqMan-等位基位-特异性扩增方法(TaqMan-ASA)应用于糖原贮积症ⅠA型中的点突变的检查。此方法是用两套等位基因特异性引物,在有TaqMan探针存在情况下,用荧光检测器对配对的PCR扩增进行实时监测。此方法是测定扩增的效率(efficiency),而不是测定终点PCR产物的存在或不存在。根据“阈值”循环确定的两种PCR扩增效率的差异来鉴别突变的和正常的等位基因,因此,使设计等位基因-特异性引物有较大的灵活性和对等位基因判断有较宽松的技术界限。此方法还可用来筛选新生儿其他遗传性代谢性疾病中的点突变。
检查基因突变的标本可用活检所得的肝或骨骼肌的新鲜标本,也可用周围血白细胞或培养的皮肤纤维母细胞。
治疗
曾采用全静脉营养疗法或鼻饲管持续点滴高碳水化合物液的治疗方案。目前主要采用每4~6小时口服生玉米淀粉(2g/kg)的替代饮食疗法,获得良好效果。
预防
1.糖原贮积症除Ⅸ型,X型,Ⅺ型三型为x连锁隐性遗传外,其余均为常染色体隐性遗传。
2.本病的发病年龄变异大,明确诊断后即应开始饮食治疗。定期测定血糖和血脂水平对监测该病的饮食治疗效果有指导价值。
3.各型GSD的临床表现虽有部分共性,但变异性很大,其分子病理学的研究和突变鉴定对临床确诊和分型有价值,但临床表型与基因型的关系的规律有待进一步评估。
4.GSD I a可通过胎儿肝活检测定G6Pase活性进行产前诊断,通常在孕18~22周进行。对已知突变的受累家庭可直接采用分子诊断技术进行产前诊断。国内已有采用分子诊断技术进行产前诊断的报道。
5.经长期有效和合理的饮食治疗可使幼年发病的女性患者正常发育,并有正常生育的能力。






























